人工知能(AI)、ビッグデータ法務 ベンチャー企業法務 ヘルスケア スタートアップ
【AI医療機器開発連載・第4回】AI医療機器開発に関する臨床研究・医学系研究関連規制の内容(全体編)

Contents
- 1 1 はじめに
- 2 2 生命・医学系研究の流れ
- 3 3 ①研究計画書の作成
- 3.1 (1)法律上適法な研究スキームの検討
- 3.2 (2)研究計画書の記載事項
- 3.2.1 ア ②研究の実施体制
- 3.2.2 イ③ 研究の目的及び意義④ 研究の方法及び期間
- 3.2.3 ウ ⑦ 第8の規定によるインフォームド・コンセントを受ける手続等
- 3.2.4 エ ⑧ 個人情報等の取扱い
- 3.2.5 オ ⑩ 試料・情報(研究に用いられる情報に係る資料を含む。)の保管及び廃棄の方法
- 3.2.6 カ ⑫ 研究の資金源その他の研究機関の研究に係る利益相反及び個人の収益その他の研究者等の研究に係る利益相反に関する状況
- 3.2.7 キ ⑬ 研究に関する情報公開の方法
- 3.2.8 ク インフォームド・アセントを得る場合には、第9の規定による手続(説明に関する事項を含む。)
- 3.2.9 ケ ㉓ 研究に関する業務の一部を委託する場合には、当該業務内容及び委託先の監督方法
- 3.2.10 コ その他
- 4 4 ②倫理審査委員会の承認
- 5 5 ③研究機関の長による許可
- 6 6 ④事前登録
- 7 7 ⑤研究の実施
- 8 8 ⑥研究の終了
- 9 9 おわりに
1 はじめに
本記事は「【連載】法規制、契約、知的財産の観点から見るAI医療機器開発」の第4回目の記事です。
【連載】法規制、契約、知的財産の観点から見るAI医療機器開発
第1 【第1回】AI医療機器の開発からサービス提供までの流れと法規制・契約
第2 医療データ収集段階の規制と契約
1 【第2回】AI医療機器開発のための医療データ収集と個人情報保護法
2 【第3回】AI医療機器開発に関する臨床研究・医学系研究関連規制の適用
3 【第4回】AI医療機器開発に関する臨床研究・医学系研究関連規制の内容(全体編)
4 【次回以降予定】AI医療機器開発に関する臨床研究・医学系研究関連規制の内容(IC編)
第3 AI医療機器開発・ハードウェア製造段階の規制と契約
第4 治験・薬事承認・保険収載段階の規制と契約
第5 サービス提供段階の規制と契約
本記事では、AI医療機器開発に関する臨床研究・医学系研究関連規制の内容をテーマに解説します。連載第1回目の記事の図でいうと、本記事のテーマは以下の箇所に該当します。
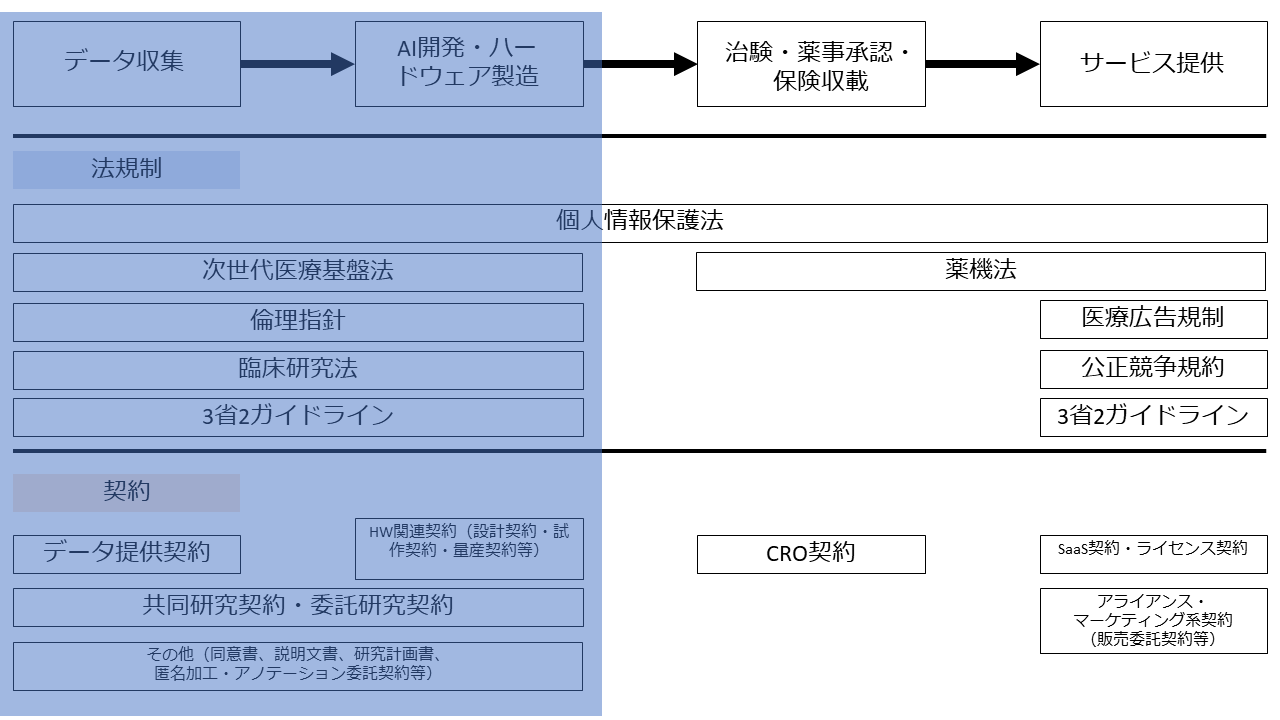
前回の「AI医療機器開発に関する臨床研究・医学系研究関連規制の適用」では、医療機器メーカーが、病院などの医療機関から患者の医療データの提供を受けてAI医療機器開発を行う場面を主に想定しつつ、「AI医療機器開発に臨床研究・医学系研究関連規制が適用されるのかどうか」という問題(研究規制の適用の有無の問題)の観点から解説しました。
今回は、「AI医療機器開発に臨床研究・医学系研究関連規制が適用されるとして、医療機器メーカーは何をしなければならないのか」という問題(研究規制の具体的な内容)について取り扱います。
AI医療機器開発の場面では多くの場合、「臨床研究法」よりも「人を対象とする生命科学・医学系研究に関する倫理指針」(以下「生命・医学系指針」といいます。)が適用されるケースが多いため、本稿ではより関心が高いと思われる生命・医学系指針に絞って解説します。
なお、生命・医学系指針におけるインフォームド・コンセントに関する規制は非常に重要なのですが、かなり複雑な規制ですので、分量の関係から本稿では取り扱わないこととし、次回以降に解説したいと思います。
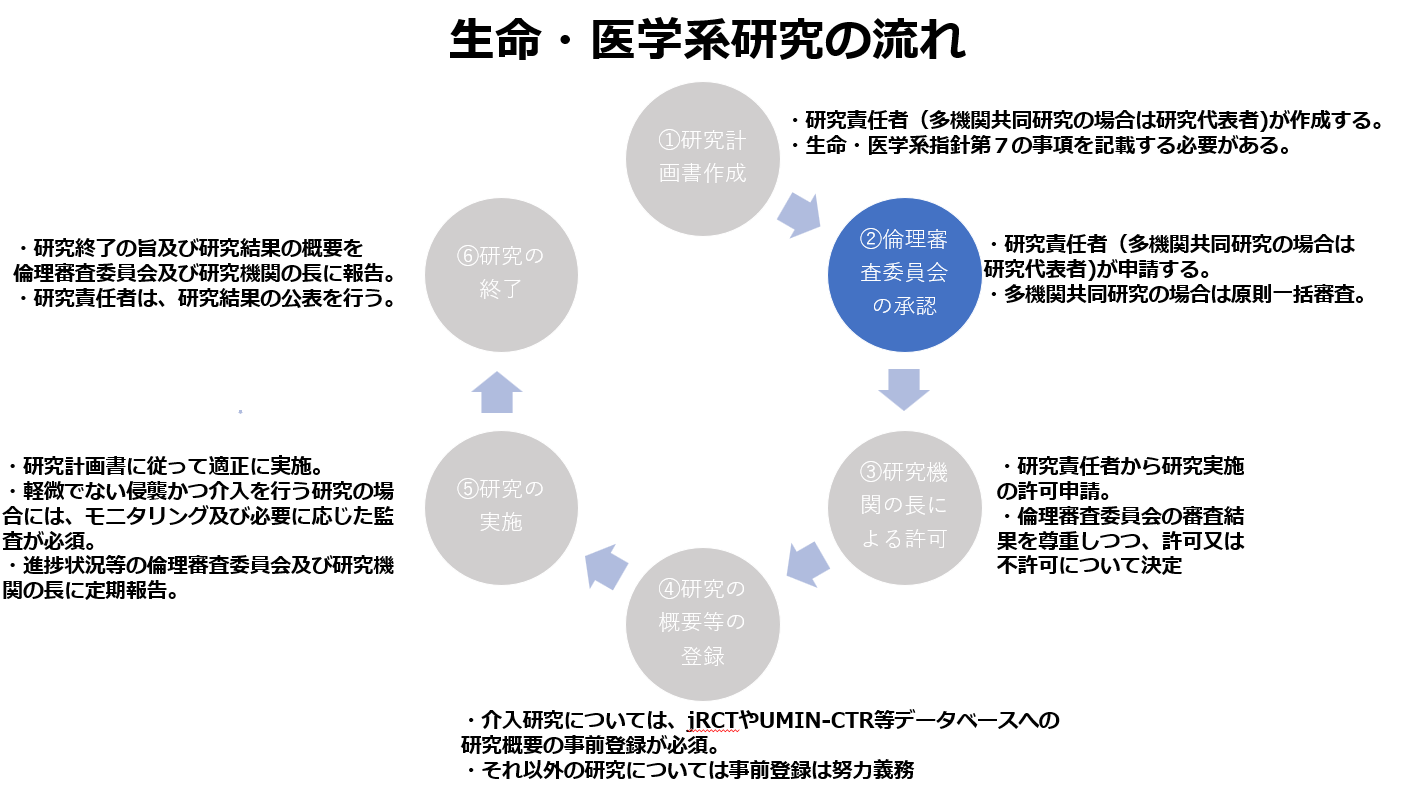
2 生命・医学系研究の流れ
生命・医学系指針の内容を確認する上では、以下の図に示される生命・医学系研究の流れを念頭に置くことが有益です。
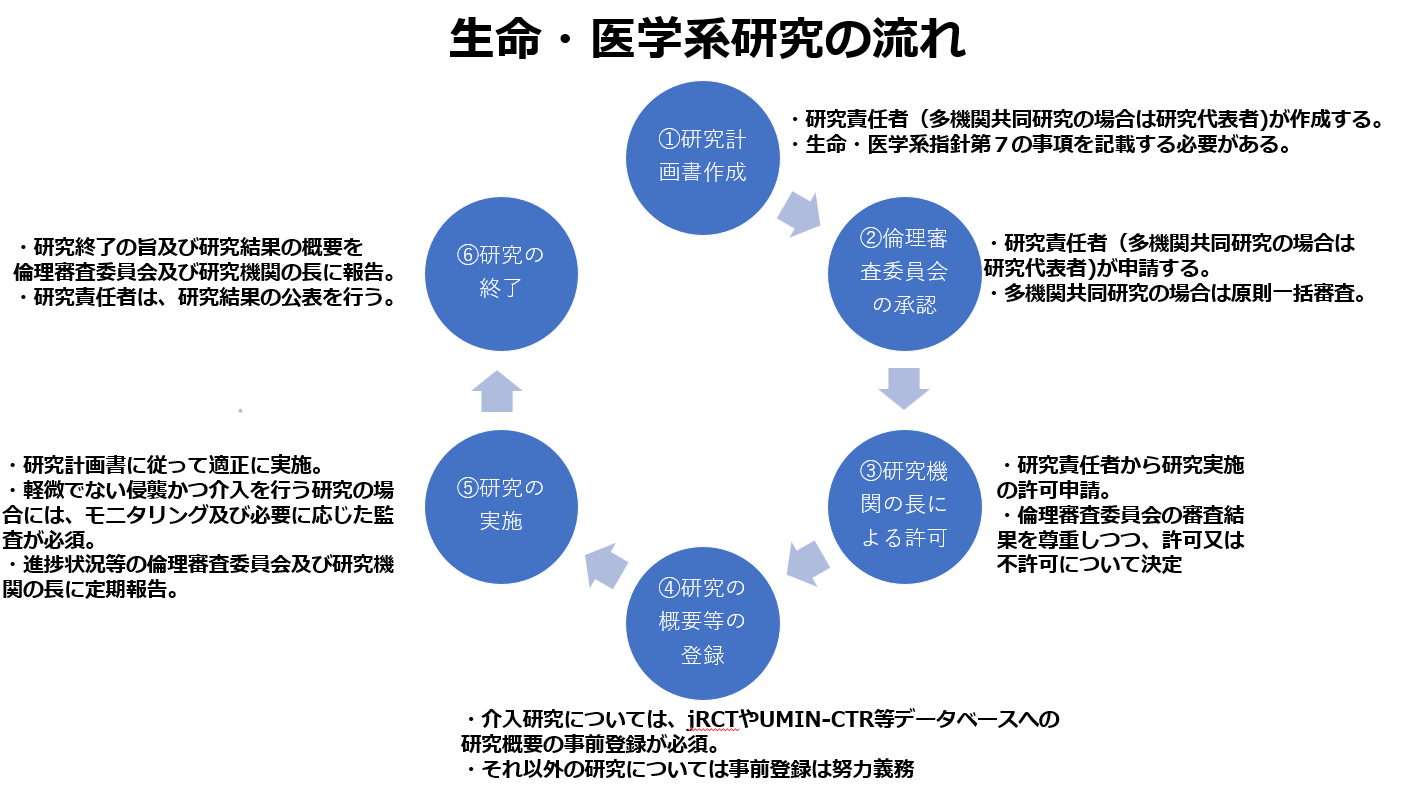
本稿では、上記の各段階において、実務上よくご相談を受ける点について解説します。
3 ①研究計画書の作成
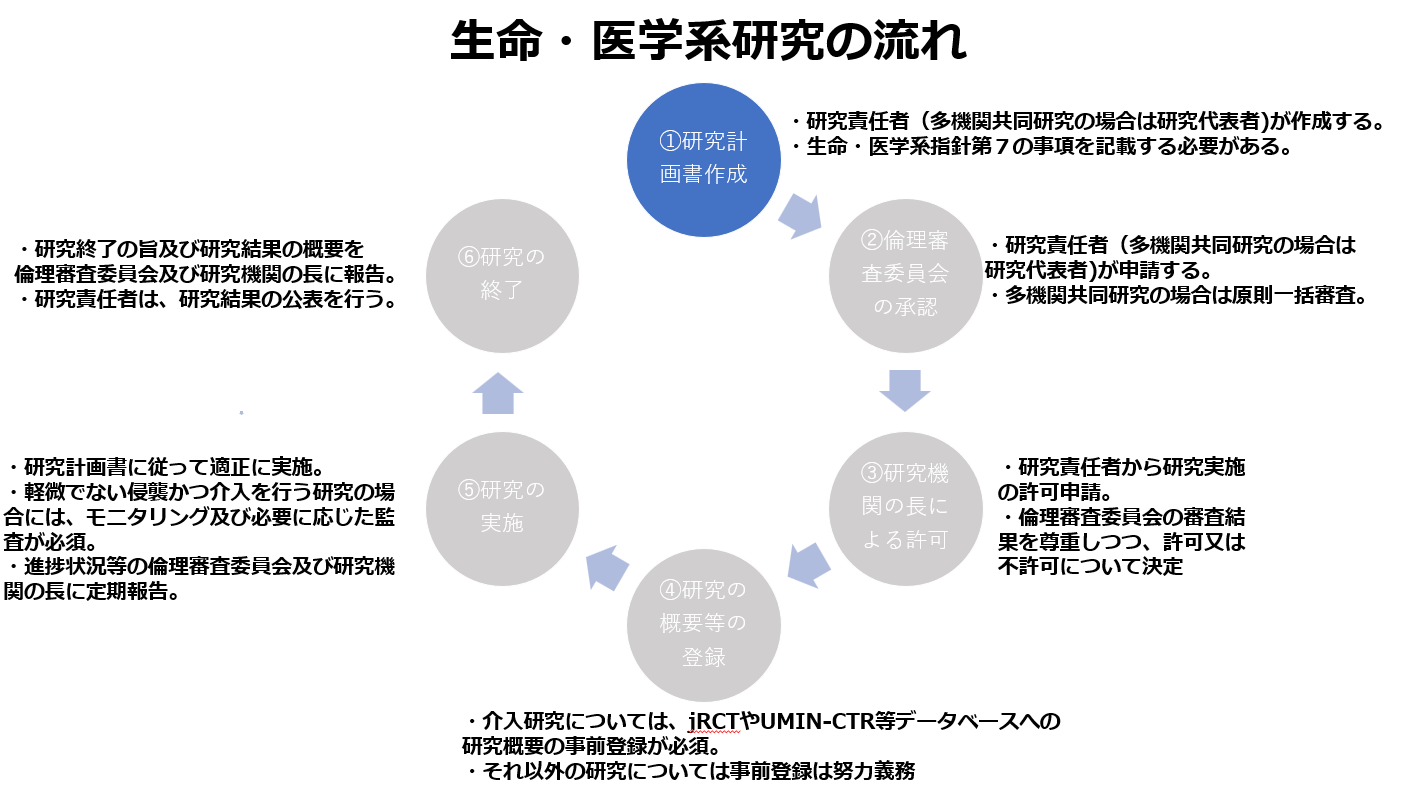
(1)法律上適法な研究スキームの検討
研究計画書を作成する前に、前提となる研究スキームを検討する必要がありますが、生命・医学系指針を遵守する以前の問題として、当然のことながら法律上適法な研究スキームである必要があります。
代表的には個人情報保護法や薬機法といった法律がよく問題になりますので、以下で少し言及します。
ア 個人情報保護法について
個人情報保護法の点については、本連載の「【AI医療機器開発連載・第2回】AI医療機器開発のための医療データ収集と個人情報保護法」をご参照ください。
イ 薬機法について
一方で、医療機器メーカーから研究スキームの検討段階でご相談を受ける際に、「開発中のAI医療機器を医療機関に使ってもらって、医師からフィードバックを得たり、利用実績を作りたい」といったご意向をお聞きすることがありますが、この点については薬機法上の規制に留意する必要があります。
前提として、薬機法上、医療機器プログラムを製造販売(医療機関に対してオンプレミスで提供することや、クラウドサービスとして電気通信回線を通じて提供することのいずれも含みます。)するためには、厚生労働大臣の「承認」又は「認証」が必要です(薬機法23条の2の5及び23条の2の23)。
そして、第3回連載でも記載したとおり、実用に耐えない精度の開発段階のAIモデルであっても「医療機器」に該当する可能性があるところ、その場合、開発中である以上は当該AIモデル(医療機器)は未承認の状態ですので、当該AIモデルを医療機関に対して提供することは、“未承認の医療機器を提供(製造販売)している”ということになり、薬機法(23条の2の5や23条の2の23)に違反する可能性があります。
この点については、厚生労働省の通達「臨床研究において使用される未承認の医薬品、医療機器及び再生医療等製品の提供等に係る医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律の適用について」があり、同通知の「3」に「未承認医薬品等の提供等に医薬品医療機器等法が適用されない場合の妥当な臨床研究」に該当するための要件として(1)~(5)の5つが記載されています。
したがって、同通達の中の5つの要件を全て満たす研究であれば、当該研究には薬機法が適用されませんので、未承認の医療機器の提供を適法に行うことができます。
ただ、5つの要件のうち、特に以下に引用する(2)の要件を満たすことが難しい場合が想定されます。
(2)医師又は歯科医師が主体的に実施する臨床研究であること。
なお、ここで主体的に実施とは、医師又は歯科医師自らが臨床研究の計画を立案し、企業等は医師又は歯科医師の求めに応じて未承認医療機器を提供等することであり、かつ、未承認医療機器に関する必要な情報は、医師又は歯科医師の求めに応じて提供することをいう。医師又は歯科医師が責任主体となっていない場合、臨床研究用とされる一連の提供行為の正当性を担保することが困難となること
つまり、多くのAI医療機器開発においては、実態としては医療機器メーカーが主体で行っていると思われ、上記(2)の要件の「医師又は歯科医師が主体的に実施する臨床研究である」と整理できるケースは限定的であると思われます。
では上記(2)の要件を満たさない場合に、医療機関に対して未承認のAI医療機器の判定結果を見せて医師からフィードバック等を得たい場合にどうすればよいのでしょうか。
対応方法としては、厚生労働省通知「医療機器プログラムの取扱いについて」に基づく対応が考えられます。
同通知の「4 電気通信回線を通じた医療機器プログラムの提供について」に以下の記載があります。
利用者から提供されたデータを使用して診断等を行うサービスは、利用者はデータの提供のみを行い、医療機器プログラムを使用しないため、医療機器プログラムの電気通信回線を通じた提供と解されないこと。ただし、電気通信回線を通じて利用者が医療機器プログラムを操作し、利用者が提供するデータから自動的に診断等の結果が提供される場合等においては、医療機器プログラムの電気通信回線を通じた提供と解される場合があることに留意すること。
要するに、医療機器プログラムを利用者に直接操作させずに、医療機器メーカーが入力データの提供を受けて判定結果だけ返すサービスであれば、当該医療機器プログラムを電気通信回線を通じて提供(製造販売)していることにはならないという意味です。
そのため、「医療機関に対して未承認のAI医療機器の判定結果を見せて医師からフィードバック等を得たい」ということであれば、真正面から未承認のAI医療機器の「提供」として整理するのではなく、同通知の考え方を前提として、「未承認AI医療機器を操作し解析しているのは医療機器メーカーであって、医療機器メーカーから医療機関に対して、解析結果を渡しているだけ」という整理も選択肢としてあり得ます。
(2)研究計画書の記載事項
上記に例示される留意点を踏まえて、法律上適法な研究スキームが固まった後は、研究計画書を作成します。
生命・医学系指針第7の(1)では、研究計画書の記載事項が定められており、①~⑮は原則的に記載が必要とされ、⑯~㉕は該当する場合のみ記載すれば良いとされています。ただし、倫理審査委員会の意見を受けて研究機関の長が許可した事項については省略することも可能です。
① 研究の名称
② 研究の実施体制(全ての研究機関及び研究協力機関の名称、研究者等の氏名並びに
既存試料・情報の提供のみを行う者の氏名及び所属する機関の名称を含む。)
③ 研究の目的及び意義
④ 研究の方法及び期間
⑤ 研究対象者の選定方針
⑥ 研究の科学的合理性の根拠
⑦ 第8の規定によるインフォームド・コンセントを受ける手続等(インフォームド・
コンセントを受ける場合には、同規定による説明及び同意に関する事項を含む。)
⑧ 個人情報等の取扱い(加工する場合にはその方法、仮名加工情報又は匿名加工情報
を作成する場合にはその旨を含む。)
⑨ 研究対象者に生じる負担並びに予測されるリスク及び利益、これらの総合的評価
並びに当該負担及びリスクを最小化する対策
⑩ 試料・情報(研究に用いられる情報に係る資料を含む。)の保管及び廃棄の方法
⑪ 研究機関の長への報告内容及び方法
⑫ 研究の資金源その他の研究機関の研究に係る利益相反及び個人の収益その他の研
究者等の研究に係る利益相反に関する状況
⑬ 研究に関する情報公開の方法
⑭ 研究により得られた結果等の取扱い
⑮ 研究対象者等及びその関係者が研究に係る相談を行うことができる体制及び相談
窓口(遺伝カウンセリングを含む。)
⑯ 代諾者等からインフォームド・コンセントを受ける場合には、第9の規定による手
続(第8及び第9の規定による代諾者等の選定方針並びに説明及び同意に関する事
項を含む。)
⑰ インフォームド・アセントを得る場合には、第9の規定による手続(説明に関する
事項を含む。)
⑱ 第8の7の規定による研究を実施しようとする場合には、同規定に掲げる全ての
要件を満たしていることについて判断する方法
⑲ 研究対象者等に経済的負担又は謝礼がある場合には、その旨及びその内容
⑳ 侵襲を伴う研究の場合には、重篤な有害事象が発生した際の対応
㉑ 侵襲を伴う研究の場合には、当該研究によって生じた健康被害に対する補償の有無及びその内容
㉒ 通常の診療を超える医療行為を伴う研究の場合には、研究対象者への研究実施後における医療の提供に関する対応
㉓ 研究に関する業務の一部を委託する場合には、当該業務内容及び委託先の監督方法
㉔ 研究対象者から取得された試料・情報について、研究対象者等から同意を受ける時点では特定されない将来の研究のために用いられる可能性又は他の研究機関に提供する可能性がある場合には、その旨、同意を受ける時点において想定される内容並びに実施される研究及び提供先となる研究機関に関する情報を研究対象者等が確認する方法
㉕ 第 14 の規定によるモニタリング及び監査を実施する場合には、その実施体制及び実施手順
以下では特に実務上よくご相談を受ける点について解説しますが、以下では「研究機関」、「共同研究機関」、「研究協力機関」、「研究者等」、「研究責任者」、「研究代表者」、「研究機関の長」といった用語が登場しますので、生命・医学系指針上の定義を引用しておきます。
なお「研究代表機関」という文言は研究の場面ではよく使われますが、生命・医学系指針上には定義規定はありません(「研究代表者」しか定義がありません)。しかし一般的な用語であると考えますので、以下本稿では研究代表者が所属する機関のことを「研究代表機関」と記載します。
⑾ 研究機関
研究が実施される法人若しくは行政機関又は研究を実施する個人事業主をいう。ただし、試料・情報の保管、統計処理その他の研究に関する業務の一部についてのみ委託を受けて行われる場合を除く。
⑿ 共同研究機関
研究計画書に基づいて共同して研究が実施される研究機関(当該研究のために研究対象者から新たに試料・情報を取得し、他の研究機関に提供を行う研究機関を含む。)をいう。
⒀ 研究協力機関
研究計画書に基づいて研究が実施される研究機関以外であって、当該研究のために研究対象者から新たに試料・情報を取得し(侵襲(軽微な侵襲を除く。)を伴う試料の取得は除く。)、研究機関に提供のみを行う機関をいう。
⒄ 研究者等
研究責任者その他の研究の実施(試料・情報の収集・提供を行う機関における業務の実施を含む。)に携わる者をいう。ただし、研究機関に所属する者以外であって、次に掲げるいずれかの者は除く。
① 新たに試料・情報を取得し、研究機関に提供のみを行う者
② 既存試料・情報の提供のみを行う者
③ 委託を受けて研究に関する業務の一部についてのみ従事する者
⒅ 研究責任者
研究の実施に携わるとともに、所属する研究機関において当該研究に係る業務を統括する者をいう。
⒆ 研究代表者
多機関共同研究を実施する場合に、複数の研究機関の研究責任者を代表する研究責任者をいう。
⒇ 研究機関の長
研究が実施される法人の代表者若しくは行政機関の長又は研究を実施する個人事業主をいう。
ア ②研究の実施体制
(ア)記載事項の留意点
「人を対象とする生命科学・医学系研究に関する倫理指針 ガイダンス」(以下「ガイダンス」といいます)のP65では、「全ての研究機関及び研究協力機関の名称、研究者等の氏名並びに既存試料・情報の提供のみを行う者の氏名及び所属する機関の名称を含む。」とされていますので、研究機関ではない、「研究協力機関」等の名称も記載する必要があります。さらに、研究機関(医療機器メーカー)に所属する「研究者等」(そのAI開発に関与する医療機器メーカーの従業員)の氏名も個別に記載する必要があります。
また、多機関共同研究の場合(例えば、医療機器メーカーも医療機関も両方「研究機関」となる場合等)には各共同研究機関の研究責任者の中から研究代表者を選任しなければならないのですが(生命・医学系指針第6 (3)。実際には医療機関側の医師に研究代表者になっていただくことが多いと思われます)、「研究代表者や各研究機関における研究責任者の役割及び責任(第6の1⑷参照)を明確に記載する必要がある。」とされており(ガイダンスP65)、この役割等が記載されていない研究計画書を見かけることもあるのでご注意ください。
(イ)医療機器メーカー及び医療機関の研究計画書上の位置づけについて
本稿では医療機器メーカーが、病院などの医療機関から患者の医療データの提供を受けてAI医療機器開発を行う場面を想定していますが、その場合の研究の実施体制については、以下のパターンが考えられます。
(ⅰ) 医療機器メーカーについて
まず、医療機器メーカーはAI医療機器を主体的に開発する立場にありますので、基本的には医療機器メーカーは「研究機関」になります。
(ⅱ) 医療機関について
一方で、医療機関を研究計画書上で「研究機関」、「研究協力機関」、「既存試料・情報の提供のみを行う者(が所属する機関)」のいずれで整理するかは、研究のスキーム(前向き研究か、後ろ向き研究か)によって複数のパターンが考えられるところです。
医療機関が単に医療機器メーカーに対して医療データを提供するだけなのであれば、前向き研究の場合には「研究協力機関」(パターンB)、後ろ向き研究の場合には「既存試料・情報の提供のみを行う者(が所属する機関)」(パターンD)として整理することも可能です。
このような、パターンBやパターンDの場合、医療機関は「研究機関」ではなく、当該医療機関に所属する医師らは「研究者等」ではありませんので、生命・医学系指針において定められた研究者等の責務の規定(第2章の第4。教育・研修を受ける義務等)や、研究機関の長の責務の規定(第2章の第5。総括的な監督や、研究の実施のための体制や規程の整備等)の適用を受けず、生命・医学系指針上の負担は比較的軽い状況で研究(AI医療機器開発)に参加・協力することができます。
ただし、特に前向き研究の場面では、パターンBではなくパターンAを検討すべき場面が考えられますので以下ご説明します。
前提として、前向き研究(医療機関において新たに医療データを取得してAI医療機器開発に用いる研究)の場合には、患者から医療情報を取得する際に、ICや適切な同意(以下「IC等」といいます。なお「IC」と「適切な同意」の違いについては次回の連載で解説します)を受ける必要がある場合があります(生命・医学系指針第4章第8の 1(1))。この点、生命・医学系指針第4章第8の 1(1)では「研究者等は、研究協力機関を介して当該研究のために新たに試料・情報を取得する場合においても、自らア又はイの手続を行う必要がある。」とされています。
そうすると、仮にパターンBの研究実施体制とした場合、IC等が必要な場合には、研究協力機関の(研究者等ではない)医師にIC等の手続を任せることができず、研究機関である医療機器メーカーの従業員(=研究者等)自身が直接IC等の手続をする必要があります。
しかし、当該研究の期間中に医療機器メーカーの従業員(研究者等)が医療機関に常駐することは現実的ではありません。
この点、生命科医学系指針第4章第8の「2 電磁的方法によるインフォームド・コンセントの取得」に基づけば、「電気通信回線を通じたテレビ電話等での対面」でIC等の手続を行うことは可能ではあるので、パターンBと整理しつつ、医療機器メーカーが遠隔でIC等の手続を行ういう選択肢もなくはありません。ただ、そのような対応をとることが難しく「医療機関の医師においてIC等の手続を行ってほしい」と考える場合には、研究の実施体制として、パターンBではなくパターンAを採用すべきと考えます。
イ③ 研究の目的及び意義④ 研究の方法及び期間
この項目は、倫理審査委員会の審査の対象となる研究の範囲(スコープ)を画する項目ですので、想定している研究について漏れなく記載する必要があります。
倫理審査委員会の承認後の研究実施段階で研究計画書に記載の無い研究を行うと、倫理審査委員会の承認の範囲を超えた研究となってしまいます。当然、AI医療機器開発においては、当然AI医療機器のアルゴリズムやプログラムの開発工程を念頭においた記載が必要です。
注意すべき事例としては、例えば以下のような事例が考えられます。
【事例1】
医療機器メーカーAが、医療機関Bから、既にBが保有している医療情報の提供を受けてAI医療機器の開発(後ろ向き研究)を行うこととし、研究計画書を作成して、倫理審査委員会の承認を得た。その後、開発は順調に進み、当該AI医療機器の薬事承認申請を行うこととなったが、申請手続における性能評価試験についても、Bから受領するデータを用いようとした。しかし、計画書内の「研究の目的及び意義」「 研究の方法及び期間」の項目には、アルゴリズムやプログラムの開発工程を念頭においた記載はあるものの、性能評価試験への使用を想定した記載は全くなかった。
前提として、厚生労働省通知「追加的な侵襲・介入を伴わない既存の医用画像データ等を用いた診断用医療機器の性能評価試験の取扱いについて」によれば、「追加的な侵襲・介入(診断結果の伝達を含む。)を伴うことなく、既存の医用画像データ又は生体試料及びこれらに関連する既存の診療情報等(通常の診療で得られたもの又はそれらを収集したバイオバンク、データベース等において提供されているものに限り、介入を伴う臨床研究等で得られたデータ等を除く。)を収集して実施する性能評価試験」は、治験には該当せずいわゆるGCP省令は適用されません。
一方で、同通知の中では「人を対象とする生命科学・医学系研究に関する倫理指針(令和3年、文部科学省・厚生労働省・経済産業省告示第1号)等を参照すること。」との記載があることからすると、通常診療で得られた医療データを用いた性能評価試験には、生命・医学系指針の適用があることになります。
そうすると、「通常診療で得られた医療データを用いた性能評価試験」を行う場合には、当該試験について研究計画書を作成して、倫理審査委員会の承認を得る必要があります。
この点、上記事例においては、「アルゴリズムやプログラムの開発工程を念頭においた記載はあるものの、性能評価試験への使用を想定した記載は全くなかった」という設定ですので、もし医療機関Bが通常診療で得た医療データを用いて医療機器メーカーAにおいて性能評価試験を行うのであれば、別途研究計画書を作成して(あるいは元の研究計画書を変更して)、倫理審査委員会の承認を得る必要があると考えます。
ただ、このような研究計画書の新規作成や変更を行ってもう一度倫理審査委員会の承認を得るというのは手間も時間もかかりますので、もし最初から性能評価試験まで見据えるのであれば、当初に作成する研究計画書の中に、アルゴリズムやプログラムの開発工程だけでなく、薬事承認における性能評価試験についても念頭においた記載にしておくべきです。
このような意味でも、「研究の目的及び意義」「 研究の方法及び期間」は慎重に検討すべき項目と言えます。
ウ ⑦ 第8の規定によるインフォームド・コンセントを受ける手続等
詳細は次回以降の記事で解説しますが、どのようにIC等の手続を行うかについても記載する必要があります。
文書によるIC等を受ける場合には、当該文書についても研究計画書に添付し、倫理審査委員会の審査を受ける必要があります。
エ ⑧ 個人情報等の取扱い
AI医療機器開発に用いる医療データに何らかの加工をしたり、個情法上の「仮名加工情報」や「匿名加工情報」を作成する場合には、その時期と方法を記載する必要があります。
なお、以前の倫理指針にはあった「匿名化」という文言は、令和4年の指針改正によって削除されていますので1「人を対象とする生命科学・医学系研究に関する倫理指針」の一部改正について(https://www.mext.go.jp/b_menu/houdou/mext_00950.html)、仮名加工情報や匿名加工情報を作成するのであれば、研究計画書内では「匿名化」という文言を使わない方が無難です。
また、匿名加工情報を作成する前提であるにもかかわらず、対応表を作成する旨の記載がある研究計画書を見かけることがありますが、「「個人情報の保護に関する法律についてのガイドライン」に関するQ&A」Q15-14を前提とすると、匿名加工情報を作成するのであれば対応表の破棄は必須となりますのでご留意ください。
オ ⑩ 試料・情報(研究に用いられる情報に係る資料を含む。)の保管及び廃棄の方法
試料・情報のトレーサビリティの観点から、保管期間を含めて記載する必要があります。
この点、「研究終了後5年間保管し、その後廃棄する」という記載を見かけることがありますが、生命・医学系指針の「第 13 研究に係る試料及び情報等の保管 」には以下の記載があります。
研究機関の長は、当該研究機関において保管する情報等について、可能な限り長期間保管されるよう努めなければならず、侵襲(軽微な侵襲を除く。)を伴う研究であって介入を行うものを実施する場合には、少なくとも、当該研究の終了について報告された日から5年を経過した日又は当該研究の結果の最終の公表について報告された日から3年を経過した日のいずれか遅い日までの期間、適切に保管されるよう必要な監督を行わなければならない。
このように、生命・医学系指針では、「可能な限り長期間保管されるよう努めなければならず」とされており、「侵襲(軽微な侵襲を除く。)を伴う研究であって介入を行うものを実施する場合」には保管期間の最低期間が定められているのみであって、保管期間が長期間にわたる場合には特に生命・医学系指針上の問題はありません。つまり、研究に用いた医療データを急いで廃棄する必要はないということです。
生命・医学系指針では以上の規制しかないにもかかわらず、より厳格な内容として「研究終了後5年間保管し、その後廃棄する」と記載してしまうと、当該研究計画書に従って廃棄する必要が生じます。
しかし、そのように記載すると、以下のような事例で不都合が生じる可能性があります。
【事例2】
医療機器メーカーAが、医療機関Bから、医療情報を匿名加工情報に加工して提供を受け、AI医療機器の開発を行った。当該開発の終了から5年後、Aは当該匿名加工情報を別のAI医療機器開発に用いることとし、当該匿名加工情報は生命・医学系指針第3・1・ウ③の「既に作成されている匿名加工情報」に該当するものとして、生命・医学系指針の適用がなく倫理審査も不要との前提で当該別のAI医療機器開発を進めようとした。しかしながら、当初のAI医療機器開発の研究計画書を確認したところ、開発に用いる医療データについて「研究終了後5年間保管し、その後廃棄する」との記載があった。
したがって、研究計画書の記載としては、例えば「少なくとも研究の中止又は終了後5年が経過した日まで保管する」・「廃棄する場合には、~という方法で行う」等と、具体的な保管期間を明記せずに最低期間を記載する方法で記載した方が、医療機器メーカーにとっての選択肢が増えて良い場合が多いと考えられます。
もちろん、もはや医療機器メーカーにおいて当該医療データを使用する見込みがないのであれば、個人情報保護法上の消去の努力義務や(個情法22条)、漏えいした場合のリスクの観点から、研究計画書上に記載された最低保管期間を経過すれば速やかに廃棄又は削除等の対応を行うのが望ましいと考えます。
カ ⑫ 研究の資金源その他の研究機関の研究に係る利益相反及び個人の収益その他の研究者等の研究に係る利益相反に関する状況
いわゆる利益相反(COI)とは、研究を適正に実施するという研究者としての使命・責務と、研究者個人の利益が衝突する状態をいいます。
一般論としては、研究者は客観的な立場から科学的に合理性をもって研究を実施する必要がありますが、もし企業から研究費の提供を受けている場合に企業に有利な結果を出した場合には、研究者の使命・責務に反することになります。
この点、利益相反があること自体が問題というわけではなく、利益相反が研究不正につながらないように管理することが重要とされています。
ガイダンスP141では、以下の記載があります。
3 ⑵の規定に関して、研究責任者は研究機関の利益相反に関する状況についての研究者等 からの報告の他、当該研究の資金源等の研究機関の研究に係る利益相反に関する状況も含めて把握し、研究計画書に記載する必要がある。
そのため、例えば、医療機器メーカーから、「共同研究機関」や「研究協力機関」である医療機関に対して研究経費が支払われる場合には、利益相反があることになりますので、その旨を研究計画書に記載する必要があります。
なお、倫理審査委員会を設置する医療機関によっては利益相反申告書を定めたり、(倫理審査委員会とは別に)利益相反委員会を設置しているものもあります。これはガイダンスP141で「利益相反委員会を設置している機関においては、研究の利益相反に関する状況について利益相反委員会の意見を求めることでも良い。」とされていることから、各医療機関の判断や内規に基づいて独自に行われているものです。
このように本来的には生命・医学系指針の上は利益相反委員会への報告は必須の対応ではないもの、医療機関側で利益相反委員会が設置されている場合には、当該医療機関の倫理審査委員会の指示に従うようにしてください。
キ ⑬ 研究に関する情報公開の方法
「6 ④事前登録」で後述します。
ク インフォームド・アセントを得る場合には、第9の規定による手続(説明に関する事項を含む。)
「中学生等の過程を未終了であり、且つ16歳未満の未成年」であって、「自らの意向を表することができると判断される場合」に「インフォームド・アセント」は取得するように努める必要がありますが、あくまでも「努力義務」です。
インフォームドコンセントとインフォームドアセントの関係性については、ガイダンスP129の以下の表がわかりやすいので以下に引用します。

ケ ㉓ 研究に関する業務の一部を委託する場合には、当該業務内容及び委託先の監督方法
解析業務や研究支援業務を外部に委託したり、共同研究機関以外の医療機関の医師からフィードバック等を受けたりするのであれば、「研究に関する業務の一部を委託する場合」に該当しますので、研究計画書内に記載する必要があります。
この点、生命・医学系指針の第6の1では以下のとおり記載されています。
⑸ 研究責任者は、研究に関する業務の一部について委託しようとする場合には、当該委託業務の内容を定めた上で研究計画書を作成又は変更しなければならない。
この部分を後から変更追記する場合にも、倫理審査委員会の承認が必要になりますので、二度手間になることを防ぐためにも、あらかじめもれなく記載しておくように留意すべきと考えます。
なお、ガイダンスP54の以下の記載を前提とすると、外部と業務委託契約を締結する場合には、必ずしも研究責任者が代表して締結する必要はなく、また多機関共同共同研究の場合には、必ずしも研究代表者が代表して締結する必要はありません。
契約の事務手続については、必ずしも研究責任者自身が行う必要はないが、研究責任者以外の者が契約の事務手続を行う場合においては、契約内容等について必ず研究責任者が責任を持って確認する必要がある。なお、多機関共同研究における業務委託については、必ずしも研究代表者が代表して締結する必要はなく、必要に応じて各研究責任者が個別に契約を締結することとしても差し支えない。
コ その他
研究計画書内で知的財産権の帰属について言及することもあります(倫理指針ではその点についての記載は義務とはありません)が、医療機器メーカーと医療機関との共同研究契約書との矛盾が生じないように、「研究対象者には帰属せず、別途合意によって定める」といった程度で記載しておく方が無難であると考えます。
4 ②倫理審査委員会の承認
(1)AI医療機器開発を行う医療機器メーカーや医療機関以外の第三者が設置する倫理審査でも良いこと
AI医療機器開発に生命・医学系指針の適用がある場合には、当該開発(研究)の実施の適否について、倫理審査委員会の意見を聴く必要があります(生命・医学系指針第6の2)。
この点、当該AI医療機器開発を行う医療機器メーカーや、当該開発に参加・協力する医療機関に倫理審査委員会が設置されていない場合であっても、外部の機関が設置する倫理審査委員会に審査を依頼することが可能です。
実際にガイダンスp159の「4 他の研究機関が実施する研究に関する審査」に関する解説部分にも「1 第 17 の4の規定は、外部の研究機関で実施する研究の審査を受託する際の責務について定めたものである。」との記載があり、ある医学系研究において、その研究の実施機関ではない機関が設置する倫理審査委員会での審査が想定されています。
(2)厚生労働省に届出が行われている倫理審査委員会であるかを確認すること
ガイダンスP151で、「倫理審査委員会を設置する者の責務」として、以下の記載があります。
⑶ 倫理審査委員会の設置者は、当該倫理審査委員会の運営を開始するに当たって、倫理審査委員会の組織及び運営に関する規程並びに委員名簿を倫理審査委員会報告システムにおいて公表しなければならない。
ここでいう「倫理審査委員会報告システム」とは、こちらのサイトのことです。
稀に、医療機器メーカーが審査依頼予定の倫理審査委員会が、当該サイトで公表されておらず、その医療機関が独自に設置しているだけのものであるケースがあります。
そのような独自に設置されているだけの倫理審査委員会は、生命・医学系指針「第 17 倫理審査委員会の役割・責務等」の「2 構成及び会議の成立要件等」を満たさない可能性もありますし、上記の公表義務も遵守していない委員会ですので、生命・医学系指針が適用されるAI医療機器開発の倫理審査の申請先としては適切ではありません。
厚労省のシステムに届けられていない倫理審査委員会への申請手続は、今後の学会発表やAIの薬事承認手続においても問題が生じる可能性が否定できず、控えられる方が無難であると考えます。
そのため、倫理審査委員会に承認申請をする場合には、上記厚労省サイトにて公表されているかどうかを確認するようにしてください。
(3)多機関共同研究の場合
多機関共同研究の場合、研究代表者は、原則として、多機関共同研究に係る研究計画書について、一つの倫理審査委員会による一括した審査を求めなければなりません。
例えば、医療機器メーカーと、その他複数の医療機関との共同研究としてAI医療機器開発を行う場合に、いずれかの医療機関の医師に研究代表者になってもらい、(当該医療機関が設置する、あるいは外部の)一つの倫理審査委員会でまとめて審査を受ける必要があります。逆に一括で審査していれば、研究代表者が所属する機関以外の研究機関において、再度個別に倫理審査委員会の審査をすることは不要です。
一方で、多機関共同研究に参加される(研究代表機関ではない)医療機関から、「研究代表機関で一括審査してもらうのはいいのだが、当院の内規では、研究代表機関の倫理審査委員会の承認とは別に、当院内部の倫理審査委員会の承認を重ねて取得する必要がある」というお声を聞くことがありますが、ガイダンスP57にも以下の記載がありますので、個別の倫理審査委員会による承認を重ねて取得することも特に問題ありません。
多機関共同研究として倫理審査委員会に審査を求める場合、「一の倫理審査委員会による場合」、「個別の倫理審査委員会による場合」が混在することを妨げるものではない。
5 ③研究機関の長による許可
倫理審査委員会の承認があれば直ちにAI医療機器開発(研究)を開始できるのではなく、医療機器メーカーの代表者等や医療機関の院長等(「研究機関の長」)の許可が必要です。
例えば医療機器メーカーと複数の医療機関等の多機関共同研究である場合には、各共同研究機関の研究責任者が各々の研究機関の長による当該研究の実施について許可を受ける必要があります。この場合、研究機関の長による許可を受けた研究機関から研究を開始することも可能です(ガイダンスP51)。
6 ④事前登録
研究機関の長の許可が出ればいよいよ研究を開始することになりますが、その前に各種データベースへの登録について検討する必要があります。
この登録については、介入(人の健康に関する様々な事象に影響を与える要因(健康の保持増進につながる行動及び医療における傷病の予防、診断又は治療のための投薬、検査等を含む。)の有無又は程度を制御する行為(通常の診療を超える医療行為であって、研究目的で実施するものを含む。))を伴う研究の場合には義務ですが、それ以外の研究は努力義務とされています。またデータベースとして、生命・医学系指針では以下のものが例示されています。
○ jRCT(Japan Registry of Clinical Trials)
https://jrct.niph.go.jp/
○ 大学病院医療情報ネットワーク研究センター 臨床試験登録システム(UMIN-CTR)
https://www.umin.ac.jp/ctr/index-j.htm
○ 国立保健医療科学院のホームページ
https://rctportal.niph.go.jp/
例えば前向き研究の場合に、「通常の診療では行わない検査を追加で行って医療データを取得し、AI医療機器開発に用いる」ということであれば、「介入」に該当すると考えますので、データベースへの登録は必須と考えます。
一方で、前向き研究であっても、例えば人の歩行動画を取得するような場合であれば、患者さんに診察室内等で実際に歩いてもらうことになるわけですが、「人の健康に関する様々な事象に影響を与える要因」の有無又は程度を制御しているわけではなく、通常の診療を超える医療行為もない(ただ歩いてもらっているだけ)という整理も可能であると考えます。
このように、前向き研究であっても「介入」があるかどうかは別問題ですので、個々の研究に照らして検討する必要があります。
一方で後ろ向き研究の場合には通常は介入は無いため、基本的にはデータベースへの登録は努力義務となります。
7 ⑤研究の実施
研究開始後は、当然のことながら、研究計画書に従って、AI医療機器開発(研究)を行う必要があります(生命・医学系研究第6・5)。
もし有害事象が発生すれば、研究責任者は速やかに必要な措置を講じ(指針第6・5)、倫理審査委員会及び研究機関の長への報告が必要です(指針第11・2)
また、前向き研究の場合にIC等を受けていたものの、そのIC等の撤回がある場合には、「当該撤回又は拒否の内容に従った措置を講ずるるとともに、その旨を当該研究対象者等に説明しなければならない」とされています(生命・医学系研究第8・9)
この同意の撤回等に関連してAI医療機器開発においてよく問題になるのは、以下のような事例です。
【事例3】
前向き研究として新たに医療データを患者から収集する際にIC等を受けていたが、その後ある患者からIC等の撤回があった。
しかし、その時点では、既にAI医療機器への学習工程は完了しており、当該患者の医療データを除外した学習用データをもう一度作成して再度学習を行うことは研究のスケジュール上、現実的な対応ではなかった。
AI医療機器の学習工程は、患者の医療データをそのままAIに取り込むのではなく、学習用データセットを用いてAIのパラメーターを更新する作業です。そのため、ある患者から同意の撤回等を申し出られても、AI医療機器の中に当該患者の医療データがそのまま入っているわけではないので、単純にAI医療機器の中からデータを削除すれば良いわけではない点が、AI医療機器開発における特殊性であると言えます。
一応、理屈の上では、同意の撤回等を申し出た患者の医療データを除外した学習用データセットをもう一度作り、学習工程をやり直すということも不可能ではないと思われますが、費用や時間の観点から、およそ現実的な対応ではないと考えます。
この点に関連して、生命・医学系指針第8・9では以下の記載があります。
ただし、当該措置を講ずることが困難な場合であって、当該措置を講じないことについて倫理審査委員会の意見を聴いた上で研究機関の長が許可したときは、この限りでない。この場合において、当該撤回又は拒否の内容に従った措置を講じない旨及びその理由について、研究者等が研究対象者等に説明し、理解を得るよう努めなければならない。
そのため、IC等を取得する同意書の中では、「学習に用いられた後に同意を撤回されたとしても、当該患者の医療データを除いてAI医療機器の再学習を行うことは困難である」旨を説明しておき、実際に同意の撤回があった場合には、倫理審査委員会の意見を得て、研究機関の長の許可をもらうことで、学習完了後のAI医療機器のパラメータその他プログラムやアルゴリズムを変更しないという対応もあり得ると考えます。
なおこの場合、学習工程が完了したAI医療機器については変更できないとしても、医療機器メーカーや医療機関が手元に保有している医療データの削除は可能と考えますので、その点の可否は検討すべきと考えます。
8 ⑥研究の終了

研究責任者は、AI医療機器開発(研究)を終了・中止したときは、その旨及び研究結果の概要を文書又は電磁的方法により遅滞なく倫理審査委員会及び研究機関の長に報告しなければなりません(生命・医学系指針第6・6(1))。「遅滞ない報告」とは、研究終了後3か月以内を目安とするとされています(ガイダンスP62)。
また、ガイダンスP62で「この指針においては研究の透明性を確保するという観点から、この指針が適用される全ての研究に関して、研究結果の公表を求めている。」とされているとおり、介入を伴うかどうかを問わず、AI医療機器開発という研究の結果を学会発表や論文掲載、公開データベースへの登録等の何らかの手段により「公表」する必要があります。
この公開の趣旨は「研究の透明性を確保する」(ガイダンスP62)ことにあるとされていますが、具体的にどこまでの粒度で研究結果を公表するのかという点については、生命・医学系指針には具体的な規定はありません。そのため、医療機器メーカーにとって企業秘密となるような重要な情報については、公開せずに秘匿するという判断も可能と思われます。
9 おわりに
今回はAI医療機器開発に適用される生命・医学系指針のうち、ICに関する規制以外の規制の全体像についてご紹介しましたが、次回以降では生命科学・医学系指針のICに関する規制についてご紹介する予定です。